|
||||
|
Глава 7 Строение молекул Теория типов Размышляя над строением органических соединений, Берцелиус пришел к выводу, что радикалы могут быть теми «кирпичиками», из которых построены органические соединения. Подобно тому как неорганические соединения построены из отдельных атомов, органические соединения построены из радикалов, которые, по мнению Берцелиуса, почти так же, как и отдельные атомы, «недоступны и неделимы». Берцелиус утверждал, что силы, удерживающие атомы в неорганической молекуле или в органическом радикале, имеют электрическую природу (что в конечном счете оказалось справедливым). Чтобы такие силы возникали, каждая молекула должна содержать положительно и отрицательно заряженные части, поскольку притяжение возможно только между противоположно заряженными частями. Доказать наличие положительно и отрицательно заряженных компонентов в простых неорганических соединениях типа хлорида натрия со временем, действительно, удалось (см. гл. 12). Однако распространить это на органические соединения оказалось значительно сложнее. Так, Берцелиус должен был настойчиво утверждать, что радикалы состоят только из углерода и водорода, причем углерод заряжен отрицательно, а водород — положительно. Он считал, что радикал бензоил (С7Н5О) не содержит и не может содержать кислород, который искажает действие, оказываемое этим радикалом. Берцелиус был также уверен, что замещение отрицательно заряженного компонента на положительно заряженный обязательно приведет к резкому изменению свойств соединения. Однако вскоре выяснилось, что это последнее его утверждение ошибочно. Одному из учеников Дюма (кстати сказать, восторженному стороннику Берцелиуса) Огюсту Лорану (1807—1853) удалось в 1836 г. заместить несколько атомов водорода в молекуле этилового спирта на атомы хлора, причем значительного изменения свойств соединения такое замещение не вызвало. Этот эксперимент противоречил теории Берцелиуса; хлор считался отрицательно заряженным, а водород — положительно заряженным элементом. Более того, в этом хлорированном соединении углерод должен был соединяться непосредственно с хлором, но как же это могло осуществиться, если оба атома заряжены отрицательно? Ведь одинаково заряженные атомы отталкиваются друг от друга. (В таком случае как могут удерживаться вместе два атома хлора в молекуле хлора? Все эти вопросы оставались нерешенными еще целое столетие.) Берцелиус, ставший в старости раздражительным и чрезвычайно консервативным, отказался пересмотреть свою точку зрения. Выслушав доклад Лорана, он яростно напал на исследователя. В 1839 г. Дюма сам получил соединение, в котором три атома водорода в уксусной кислоте были замещены хлором, но, боясь потерять расположение Берцелиуса, малодушно отступил и отрекся от взглядов Лорана. Из-за гнева Берцелиуса перед Лораном оказались закрытыми двери наиболее известных лабораторий, однако Лоран был настойчив и продолжал собирать доказательства того, что радикалы не являются «неразрушимыми и недоступными», как это утверждал Берцелиус, и что не следует переоценивать влияние положительного и отрицательного зарядов. Хотя химиков и одолевали сомнения, авторитет Берцелиуса был настолько велик, что вплоть до смерти этого крупнейшего ученого (1848 г.) никто не решался отступиться от его теории радикалов. Однако после смерти Берцелиуса популярность идей Лорана сразу возросла, и у него появились сторонники. Лоран отказался от всякого подчеркивания влияния электрических сил. Он полагал, что органическая молекула имеет ядро (которое может представлять собой одиночный атом), к которому присоединяются различные радикалы. Органические молекулы можно сгруппировать в семейства или типы (отсюда теория типов) [57]. Молекулы соединений одного типа должны иметь сходные ядра. К этим ядрам могут присоединяться любые из радикалов, образующих ряд подобных радикалов. Отдельные типы молекул можно распространить и на неорганические соединения. В соответствии с представлениями этой теории в молекуле воды (Н2О) к центральному атому кислорода — ядру присоединены два атома водорода. Замещая один из атомов водорода на радикалы какого-либо ряда, можно получить группу соединений, в число которых входят и вода, и различные органические соединения. При замещении одного из атомов водорода на метильную (СН3) или этильную (С2Н5) группу образуются соответственно метиловый (СН3ОН) или этиловый (С2Н5ОН) спирт. В результате такого замещения можно получить чрезвычайно много различных спиртов. И действительно, спирты не только имеют много общего между собой, но и проявляют также определенное сходство с водой. Простые спирты — метиловый и этиловый смешиваются с водой в любых соотношениях. Щелочные металлы реагируют со спиртами так же, как с водой, хотя и более медленно. Между 1850 и 1852 гг. английский химик Александр Уильям Уильямсон (1824—1904) показал, что органические соединения, относящиеся к классу простых эфиров, можно также построить по «типу воды». Простые эфиры можно получить, заместив на органические радикалы оба атома водорода воды. В обычном эфире, который в то время начали применять как анестезирующее средство, оба атома водорода замещены на этильные группы, так что его формула записывается как С2Н5ОС2Н5. Несколько ранее, в 1848 г., французский химик Шарль Адольф Вюрц (1817—1884), изучавший группу соединений, связанных с аммиаком и потому названных аминами, показал, что у соединений этого типа ядром служит атом азота. В аммиаке атом азота связан с тремя атомами водорода, в аминах один или несколько атомов водорода замещены на органические радикалы. Теория типов завоевывала все большую популярность, поскольку она позволила систематизировать органические соединения, а список все новых и новых органических соединений в то время очень быстро увеличивался. Русский химик Федор Федорович Бейльштейн (1838—1906) опубликовал в 1880 г. обширное руководство по органическим соединениям, в котором использовал теорию типов Лорана для размещения этих соединений в рациональном порядке. И тем не менее теория типов в том виде, в каком она вытекала из работ Лорана, оставалась незавершенной. По-прежнему предполагалось, что органические соединения построены из радикалов, вопрос о молекулярной структуре обходился стороной. Ответить на него можно было, только выяснив, как в действительности располагаются атомы в самих радикалах. Валентность Некоторые химики считали теорию типов весьма упрощенной. Вызывало удивление также то обстоятельство, что, согласно теории типов, кислород неизменно оказывался связанным с двумя другими атомами или радикалами, Так, в молекуле воды кислород был связан с двумя атомами водорода, в молекуле спирта — с одним атомом водорода и одним органическим радикалом, в молекуле эфира — с двумя органическими радикалами. А азот всегда соединялся с тремя атомами или радикалами. Английский химик Эдуард Франкланд (1825—1899) первым заинтересовался металлорганическими соединениями, в которых органические группировки присоединены непосредственно к атомам металла, например цинка [58]. В соединениях такого типа, как было установлено, каждый атом металла присоединяет определенное число органических групп, причем оно различно для разных металлов. Например, атомы цинка соединяются с двумя (не больше и не меньше) органическими группами. В 1852 г. Франкланд выдвинул теорию, которая позднее стала известна как теория валентности (от латинского valentia — сила) [59], согласно которой каждый атом обладает определенной способностью к насыщению (или валентностью). Так, атом водорода в нормальных условиях соединяется только с одним атомом другого типа. То же самое можно сказать о натрии, хлоре, серебре, броме и калии. Валентность всех перечисленных элементов равна единице. Атомы кислорода соединяются не менее чем с двумя различными атомами. Так же ведут себя кальций, сера, магний и барий. У этих элементов валентность два. У азота, фосфора, алюминия и золота валентность три. Железо может иметь валентность два или три. В принципе вопрос о валентности оказался не столь простым, каким представлялось вначале, но даже такой простейший вариант этой теории позволил сделать важные выводы. Прежде всего с введением понятия «валентность» удалось уяснить различие между атомным весом (см. гл. 6) и эквивалентным весом элементов. Даже в середине XIX столетия многие химики еще путали эти два понятия. При образовании хлорида водорода 1 атом водорода соединяется с 1 атомом хлора, а поскольку атом хлора в 35.5 раза тяжелее атома водорода, то, следовательно, водород и хлор соединяются в соотношении 1:35.5, т. е. атомный вес хлора равен 35.5. Однако такое соотношение элементов наблюдается не во всех соединениях. Например, каждый атом кислорода соединяется с 2 атомами водорода, так как валентность кислорода равна двум. Поскольку атомный вес кислорода равен 16, следовательно, 16 частей кислорода соединяются с 2 частями водорода. В результате эквивалентный вес кислорода, соединяющегося с 1 частью водорода, равен 16/2, или 8. Аналогично атом азота (атомный вес 14, валентность 3) соединяется с 3 атомами водорода. Следовательно, эквивалентный вес азота равен 14/3, или примерно 4.7. В общем эквивалентный вес атома равен его атомному весу, деленному на его валентность. В то же время второй закон электролиза Фарадея (см. гл. 5) гласит, что вес металла, выделяемого в свободном состоянии при прохождении данного количества электричества, пропорционален эквивалентным весам этих металлов. Это означает, что при прохождении данного количества электричества вес выделяемого в свободном состоянии двухвалентного металла составляет только половину веса выделяемого в свободном состоянии одновалентного металла примерно равного атомного веса. Это положение можно объяснить следующим образом: для перемещения одного одновалентного атома требуется один «атом электричества» (см. гл. 5), в то время как для перемещения одного двухвалентного атома требуются два «атома электричества». Однако природу зависимости между валентностью и «атомами электричества» удалось полностью выяснить лишь спустя еще полстолетия (см. гл. 5). Структурные формулы [60] Теория валентности сыграла важнейшую роль в развитии теории химии вообще и органической химии в особенности. Исходя из теории валентности, Кекуле предположил, что атом углерода четырехвалентен, и в 1858 г. попытался, опираясь на это предположение, представить строение наиболее простых органических молекул и радикалов [61]. В том же 1858 г. шотландский химик Арчибальд Скотт Купер (1831—1892) предложил изображать силы, соединяющие атомы (или связи, как их принято называть), в виде черточек. После того как была «построена» первая органическая молекула, стало совершенно ясно, почему органические молекулы, как правило, значительно больше и сложнее, чем неорганические. Согласно представлениям Кекуле, углеродные атомы могут соединяться друг с другом с помощью одной или нескольких из четырех своих валентных связей, образуя длинные цепи — прямые или разветвленные. По-видимому, никакие другие атомы не обладают этой замечательной способностью в той мере, в какой обладает ею углерод. Итак, представив себе, что у каждого атома углерода четыре валентные связи, а у каждого атома водорода одна такая связь, можно изобразить три простейших углеводорода (соединения, молекулы которых образованы только атомами углерода и водорода), метан CH4, этан C2H6 и пропан C3H8, следующим образом: 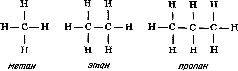 Увеличивая число атомов углерода, эту последовательность можно продолжить, причем практически бесконечно. Добавляя к углеводородной цепи кислород (две валентные связи) или азот (три валентные связи), можно представить структурные формулы молекул этилового спирта (C2H6O) и метиламина (CH5N): 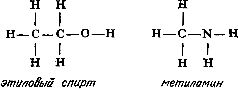 Допустив возможность наличия между соседними атомами двух связей (двойная связь) или трех связей (тройная связь), можно изобразить структурные формулы таких соединений, как этилен (C2H4), ацетилен (C2H2), метилцианид (C2H3N), ацетон (C3H6O) и уксусная кислота (C2H4O2): 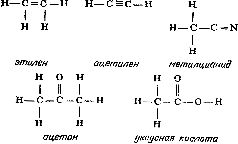 Полезность структурных формул была настолько очевидной, что многие химики-органики приняли их сразу. Они признали полностью устаревшими все попытки изображать органические молекулы как структуры, построенные из радикалов. В результате было признано необходимым, записывая формулу соединения, показывать его атомную структуру. Русский химик Александр Михайлович Бутлеров (1823—1886) использовал эту новую систему структурных формул в разработанной им теории строения органических соединений [62]. В 60-х годах прошлого столетия он показал, как с помощью структурных формул можно наглядно объяснить причины существования изомеров (см. гл. 5). Так, например, у этилового спирта и диметилового эфира одна и та же эмпирическая формула C2H6O, однако структурные формулы этих соединений значительно различаются: 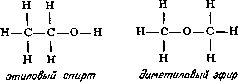 поэтому не удивительно, что изменение в расположении атомов приводит к двум наборам очень разных свойств. В этиловом спирте один из шести атомов водорода присоединен к атому кислорода, в то время как в диметиловом эфире все шесть атомов водорода присоединены к атомам углерода. Атом кислорода удерживает атом водорода слабее, чем атом углерода, так что металлический натрий, добавленный к этиловому спирту, замещает водород (одну шестую общего количества). Натрий, добавленный к диметиловому эфиру, совсем не вытесняет водород. Таким образом, при составлении структурных формул можно руководствоваться химическими реакциями, а структурные формулы, в свою очередь, могут помочь понять суть реакций. Бутлеров особенно много внимания уделил одному из типов изомерии, называемому таутомерией (динамической изомерией), при которой некоторые вещества всегда выступают как смеси двух соединений. Если одно из этих соединений выделить в чистом виде, оно сразу же частично перейдет в другое соединение. Бутлеров показал, что таутомерия обусловлена спонтанным переходом атома водорода от атома кислорода к соседнему атому углерода (и обратно). Чтобы вполне доказать справедливость системы структурных формул, необходимо было определить структурную формулу бензола — углеводорода, содержащего шесть атомов углерода и шесть атомов водорода. Сделать это удалось далеко не сразу. Казалось, не существует такой структурной формулы, которая бы, отвечая требованиям валентности, в то же время объясняла бы большую устойчивость соединения. Первые варианты структурных формул бензола очень походили на формулы некоторых углеводородов — соединений весьма нестойких и не похожих по химическим свойствам на бензол. Решить эту задачу смог опять-таки Кекуле. В один из дней 1865 г. (как он сам рассказывает) Кекуле в полудреме ехал в омнибусе, и ему пригрезилось, что он видит атомы, кружащиеся в танце. Вдруг конец одной цепи соединился с ее началом, и образовалось вращающееся кольцо. И Кекуле решил, что именно такой должна быть структурная формула бензола. До тех пор структурные формулы строились только в виде линейных цепей углеродных атомов, но теперь Кекуле ввел понятие «кольцо» (или «ядро») атомов углерода и предложил следующую структурную формулу бензола: 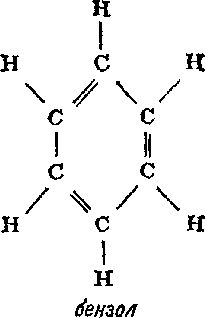 Это объяснение было принято, и представление о структурных формулах расширилось [63]. Оптические изомеры [64] Структурные формулы оказались чрезвычайно полезными, но они не отражали один особенно сложный тип изомерии — оптическую изомерию. Прежде чем перейти к этому типу изомерии, рассмотрим вкратце природу света. В 1801 г. Томас Юнг (1773—1829), выдающийся английский физик, астроном и врач (разработавший, в частности, теорию цветного зрения), провел опыты, показавшие, что свет ведет себя так, как будто он состоит из очень маленьких волн. Затем, примерно в 1814 г., французский физик Огюстен Жан Френель (1788—1827) показал, что световые волны относятся к классу волн, называемых поперечными волнами. В таких волнах колебания происходят под прямым углом к направлению их распространения. Самый наглядный пример волн такого типа — волны на воде. Отдельные частицы воды перемещаются вверх и вниз, а сама ванна движется по поверхности. Световые волны — это не волны на поверхности, а потому колебания в них не должны происходить обязательно в направлении вверх-вниз. Число направлений, в которых колебания световых волн могут происходить под прямым углом к направлению их распространения, практически бесконечно. В луче обычного света ни одно из направлений колебаний не является предпочтительным. Однако если такой луч света пропустить через некоторые кристаллы, то упорядоченное расположение атомов в кристалле заставит световые колебания происходить только в какой-то определенной плоскости — в плоскости, которая позволяет лучу проходить и обходить ряды атомов. Свет с колебаниями только в одной плоскости был назван в 1808 г. французским физиком Этьеном Луи Малюсом (1775—1812) поляризованным светом. В то время волновая теория еще не завоевала признание, и Малюс полагал, что свет состоит из частиц с северным и южным полюсами и что в поляризованном свете все полюсы ориентированы в одном направлении. Эта теория вскоре была отвергнута, но название, данное Малюсом, осталось и используется до сих пор. Первоначально свойства и поведение поляризованного света интересовали исключительно физиков. Однако в 1815 г, французский физик Жан Батист Био (1774—1862) показал, что при прохождении поляризованного света через некоторые кристаллы происходит поворот плоскости колебаний (плоскости поляризации) световых волн. В одних случаях она поворачивается по часовой стрелке (правое вращение), в других — против часовой стрелки (левое вращение). К числу кристаллов, обладающих указанным свойством,— оптической активностью, относятся и кристаллы ряда органических соединений. Более того, некоторые из этих органических соединений, например различные сахара, оптически активны и в растворах. Со временем выяснилось, что некоторые соединения отличаются друг от друга только своими оптическими свойствами. Одно из таких одинаковых по всем другим свойствам соединений вращает плоскость поляризации поляризованного света по часовой стрелке, другое — против часовой стрелки. Обычно имеется еще и третье соединение, которое вообще не вызывает вращения плоскости поляризации поляризованного света (оптически неактивно). Примером изомерных веществ, различающихся по оптической активности, могут служить открытые Берцелиусом (см. гл. 6) виноградная и винная кислоты. Виноградная кислота оптически неактивна, а винная кислота обладает в растворе правым вращением. Позднее была открыта винная кислота, обладавшая в растворе в тех же условиях равным по величине, но противоположным, левым вращением. Эти две формы винных кислот — природная правовращающая и не встречающаяся в природе левовращающая винная кислота — пример оптических изомеров. Объяснить причину возникновения изомерии только с помощью структурных формул Кекуле невозможно. Первый шаг в этом направлении был сделан в 1848 г. французским химиком Луи Пастером (1822—1895). Кристаллизуя из водного раствора винограднокис-лый натрий-аммоний при комнатной температуре, Пастер обнаружил, что образованные в этих условиях кристаллы асимметричны. Причем наблюдаются две формы кристаллов: правая и левая (при одинаковой ориентации кристаллов небольшая характерная грань у одних кристаллов находилась слева, а у других — справа). Пастер сумел под увеличительным стеклом при помощи пинцета тщательно разделить оба типа кристаллов. Свойства растворов этих кристаллов оказались полностью идентичными, исключение составляла только их оптическая активность — растворы обладали противоположным вращением. Превратив кристаллы, обладающие в растворе правым вращением, в кислоту, Пастер обнаружил, что получил известную ранее природную правовращающую винную кислоту, из кристаллов другого типа получался ее оптический изомер — ранее не известная левовращающая винная кислота. Отсюда Пастер сделал вывод, что в кристаллах виноградной кислоты содержится равное количество молекул право- и левовращающих винных кислот и именно поэтому виноградная кислота оптически неактивна. Соединения, подобные виноградной кислоте, стали называть рацемическими (от латинского названия виноградной кислоты). Результаты этих опытов убедительно свидетельствовали о том, что оптическая активность связана с асимметрией. Однако асимметрия наблюдалась у кристаллов, а многие вещества проявляли оптическую активность как в кристаллическом состоянии, так и в растворах. При растворении веществ происходит разрушение упорядоченной упаковки молекул в кристаллах, и в растворе вещества находятся в виде отдельных беспорядочно перемещающихся молекул. Если оптическая активность обусловлена асимметрией, то асимметрична должна быть и сама структура молекул. Из структурных формул не следует, что возможно существование асимметричных молекул, однако это не позволяет говорить об отсутствии связи между асимметрией и оптической активностью. Структурные формулы записываются на плоской поверхности доски или листа бумаги, но едва ли органические молекулы в действительности являются двумерными. Несомненно, молекулы трехмерны и образующие их атомы в действительности размещаются в трех измерениях. Расположив атомы таким образом, легко выявить ту самую асимметрию молекулы, которая обусловливает ее оптическую активность. Однако как представить себе, что молекула трехмерна? Атомов никто никогда не видел, и само их существование могло казаться удобной выдумкой, используемой для объяснения химических реакций. Как же располагать в пространстве то, что, возможно, и не существует? Следующий шаг мог сделать только молодой человек, еще не обремененный той мудрой осторожностью, которая приходит лишь с годами. Молекулы в трех измерениях Таким человеком оказался молодой датский химик Якоб Гендрик Вант-Гофф (1852—1911) [65]. В 1874 г., когда Вант-Гофф еще работал над докторской диссертацией, он выдвинул смелое предположение, согласно которому четыре связи углеродного атома направлены к четырем вершинам тетраэдра, в центре которого находится этот атом. Представить себе это можно так: три связи атома углерода образуют треногу, а четвертая связь направлена прямо вверх. Все четыре связи при этом равноудалены друг от друга, а угол между любыми двумя соседними связями равен примерно 109° (рис. 11).  Рис. 11. Тетраэдрическое расположение связей атомов углерода допускает две конфигурации, одна из которых является зеркальным отображением другой. На рисунке показаны два возможных варианта расположения атомов в молекуле молочной кислоты. Таким образом, четыре связи атома углерода располагаются симметрично относительно атома, и симметрия нарушается лишь в том случае, когда все четыре связи присоединяются к различным атомам или группам атомов. Поскольку присоединение может быть осуществлено двумя различными способами, полученные фигуры представляют собой зеркальные изображения друг друга. Это дает как раз тот тип асимметрии, который Пастер обнаружил в кристаллах винной кислоты. Почти одновременно с Вант-Гоффом подобные предположения опубликовал французский химик Жозеф Ашиль Ле Бель (1847—1930). Поэтому тетраэдрическую модель атома углерода иногда называют моделью Вант-Гоффа — Ле Беля. Гипотеза Вант-Гоффа — Ле Беля быстро завоевала признание. Этому, в частности, способствовала книга, выпущенная в 1887 г. немецким химиком Йоханнесом Адольфом Вислиценусом (1835—1902), который был широко известен в научном мире и пользовался большим авторитетом. Соединения с асимметрическим атомом углерода (соединенным с четырьмя разными группировками) могут существовать в виде оптически активных изомеров; соединения, не имеющие таких атомов, не проявляют оптической активности. Более того, у соединений с несколькими асимметрическими углеродами число экспериментально найденных оптически активных изомеров всегда совпадало с предсказанным на основании теории Ле Беля — Вант-Гоффа. В конце XIX столетия утвердилось мнение, что пространственное расположение связей присуще не только атому углерода. Немецкий химик Виктор Мейер (1848—1897) показал, что некоторые типы оптической изомерии, наблюдаемые у азотсодержащих соединений, можно объяснить, лишь допустив пространственное расположение связей азота. В 1900—1902 гг. английский химик Уильям Джексон Поуп (1870—1939) продемонстрировал, что трехмерную модель можно распространить также на атомы серы, селена и олова, а несколько позднее швейцарский химик Альфред Вернер (1866—1919) добавил к этому списку кобальт, хром, родий и ряд других металлов. (Начиная с 1891 г. Вернер занимался разработкой координационной теории, которая позволила бы объяснить свойства некоторых «необычных неорганических соединений». Согласно этой теории, кроме главных валентных сил имеются еще и силы побочной валентности. Первоначально считалось, что они резко отличаются от основных валентных сил, но впоследствии выяснилось, что существенного различия между ними не существует. Другим важнейшим положением теории Вернера была идея о том, что группировки, связанные с атомами металла, располагаются вокруг них в пространстве в вершинах определенных многогранников (атом металла, расположенный в центре многогранника, получил название центрального атома). Теория Вернера смогла объяснить и предсказать многочисленные случаи изомерии координационных соединений, в том числе и оптической изомерии.) С появлением трехмерной модели молекулы теория строения молекулы начала быстро развиваться. Виктор Мейер показал, что обычно группы атомов могут свободно вращаться вокруг единственной связи, соединяющей их с остальной частью молекулы, но в ряде случаев этому вращению препятствуют соседние объемные группы. Поуп продолжил эти исследования и показал, что такая пространственно затрудненная молекула в целом может оказаться асимметричной и будет проявлять оптическую активность, хотя ни один из составляющих ее атомов сам по себе не является асимметрическим. Немецкий химик Иоганн Фридрих Вильгельм Адольф фон Байер (1835—1917) использовал в 1885 г. идею трехмерного строения молекул для изображения пространственного строения циклических соединений (в виде плоских колец). Если четыре связи атомов углерода направлены к четырем углам тетраэдра, то угол между любыми двумя связями составляет 109°28'. Байер утверждал, что в любом органическом соединении атомы располагаются, как правило, так, что углы между связями атома углерода примерно соответствуют приведенному значению. Если же по какой-либо причине угол меняется, то атом оказывается в напряженном состоянии. Если три атома углерода соединены друг с другом в цикл, то они образуют равносторонний треугольник, в котором угол между каждой парой связей равен 60°, т. е. значительно отличается от естественного угла 109°28'. По этой причине циклы из трех атомов углерода образуются с трудом, а если и образуются, то легко разрушаются. Четыре атома углерода, согласно Байеру, образуют квадрат с углами 90°, пять атомов углерода образуют пятиугольник с углами 108°, а шесть атомов — шестиугольник с углами 120°. Вполне очевидно, что образование пятиугольника по существу не приводит к возникновению напряжений в связях атомов углерода, связи атомов в шестичленном кольце напряжены лишь в небольшой степени. Следовательно, с помощью теории напряжения Байера можно было по-видимому, объяснить, почему среди природных циклических соединений преобладают пяти- и шестичленные [66]. Однако решающей проверке теория Вант-Гоффа — Ле Беля подверглась в работах немецкого химика Эмиля Фишера (1852—1919), занимавшегося изучением простых сахаров. Ко времени начала работы Фишеру было известно, что ряд сахаров имеет одну и ту же эмпирическую формулу С6Н12О6 и обладает многими сходными свойствами, но различается, в частности, по оптической активности. Фишер показал, что в молекуле каждого из этих сахаров имеются четыре асимметрических атома углерода, т. е., согласно теории Вант-Гоффа — Ле Беля, они должны иметь шестнадцать оптически активных изомеров. Эти изомеры можно расположить в виде восьми пар; в каждой такой паре изомеры вращают плоскость поляризованного света на одну и ту же величину, один по часовой стрелке, а другой — против. Фишер продолжил свою работу и установил расположение заместителей у трех асимметрических атомов углерода в молекулах ряда изомерных сахаров относительно заместителей при четвертом асимметрическом углероде, пространственное расположение которых было выбрано произвольно, поскольку в то время не существовало прямых методов его определения. (Спустя шестьдесят лет было установлено, что произвольный выбор, сделанный Фишером, оказался правильным). В результате этих работ стереохимическая теория Вант-Гоффа — Ле Беля получила наглядное и весьма впечатляющее подтверждение, что окончательно убедило химиков в ее справедливости. Предсказания теории подтвердились и при изучении ряда других соединений, в частности сахаров других типов, аминокислот и пр. К 1900 г. трехмерная модель молекулы была принята практически всеми учеными [67]. Примечания:[5] «Элемент» — латинское слово неизвестного происхождения. Греки не употребляли его, но, поскольку это одно из важнейших понятий современной химии, обойтись без него невозможно, даже в тех случаях, когда речь идет о греках. [6] Очень важным для формирования качественно-химических представлений о веществе было введенное последователем Фалеса Анаксимандром (ок. 610— 546 гг. до н. э.) понятие ??????? — первоначало, порождающее бесконечное многообразие всего сущего с различными качествами. [57] О доструктурных теориях органической химии (теории радикалов, теории этерина, теории замещения Дюма, «старой» теории типов Дюма и «новой» теории типов Ш. Ф. Жерара см.: Быков Г. В. История классической теории химического строения.— М.: Изд-во АН СССР, 1960, 311 с. О вкладе Шарля Фредерика Жерара (1816—1856) в развитие теоретической органической химии см.: Фаерштейн М. Г. Шарль Жерар.— М.: Наука, 1968, 163 с. [58] В истинных металлорганических соединениях атом металла прочно связан с атомом углерода. Соединения, подобные ацетату цинка (вещества такого типа были известны и до Франкланда), являются солями органических кислот. В таких солях атом металла присоединяется к атому кислорода, и они не считаются истинными металлорганическими соединениями. [59] О возникновении учения о валентности и появлении понятия «химическая связь» см.: Быков Г. В. История органической химии. Структурная теория. Физическая органическая химия. Расчетные методы.— М.: Химия, 1976, 360 с. [60] Главные положения теории строения высказал А. М. Бутлеров в докладе «О химическом строении вещества», сделанном 9 сентября 1861 г. на съезде немецких естествоиспытателей и врачей. Бутлеровым были сформулированы правила, которыми можно было руководствоваться при определении строения органических соединений, а также было объяснено явление изомерии. А. Кекуле в 1865 г. распространил положения теории строения на ароматические соединения. Экспериментальное подтверждение теории химического строения Бутлеровым и его учениками имело огромное значение для ее утверждения.— Прим. ред. [61] Впервые А. М. Бутлеров изложил свои взгляды на теорию строения в лекциях, прочитанных им в Казанском университете в 1860 г., а в 1861 г. на Съезде немецких естествоиспытателей выступил с подробным докладом на эту тему. Самым главным вкладом Бутлерова, отличающим его труды от работ А. Кекуле и А. С. Купера, было последовательно проводимое положение о взаимосвязи между химическим строением и свойствами молекул. Это сделало понятие о химическом строении важнейшим теоретическим элементом химии. Подробнее см. упомянутые выше книги Г. В. Быкова (примечания 62 и 64). [62] См.: Быков Г. В. Александр Михайлович Бутлеров. Очерк жизни и деятельности.— М.: Изд-во АН СССР, 1961, 218 с; Быков Г. В. О приоритете А. М. Бутлерова в создании теории химического строения. В кн.: Материалы по истории отечественной химии.— М.: Изд-во АН СССР, 1953, с. 20—32. [63] Однако объяснить загадку двойных связей бензола, которые ведут себя не так, как двойные связи в других соединениях, удалось лишь спустя примерно три четверти века (см. гл. 12). [64] См.: Быков Г, В. История стереохимии органических соединений.— М.: Наука, 1966, 372 с. [65] См.: Добротин Р. Б., Соловьев Ю. И. Вант-Гофф.— М.: Наука, 1977, 272 с. [66] Теория напряжения Байера в свое время удовлетворительно объясняла нестойкость циклов малого размера (трех- и четырехчленных). Однако впоследствии было установлено, что тетраэдрические атомы углерода в циклических системах не находятся в одной плоскости, поэтому возможно построение шестичленных циклов и любых циклов большего размера, свободных от углового напряжения. [67] По материалам этой главы см. также: Фаерштейн М. Г. История учения о молекуле в химии (до 1860 г.).— М.: Изд-во АН СССР, 1961, 368 с. |
|
|||
|
Главная | В избранное | Наш E-MAIL | Добавить материал | Нашёл ошибку | Наверх |
||||
|
|
||||